
|
2018 Pharmaceutical |
||||
|
Transfer of a validated HPLC method to quantify related-substances in API (Active Pharmaceutical Ingredient) Techer Alexandre |
||||
|
Introduction The quality of an API is essential because it will be consumed by sick persons. That is why we have to identify and quantify these eventual impurities localized inside the API. FDA (Food and Drug Agency) pharmacopeia for example decrees that pharmaceutical companies, which sell medicines in the USA, must abide by several specifications about the quality of the health products. Siegfried Saint Vulbass site produces API for different customer including American customers. A new product is manufacturing here and the quality control laboratory has to transfer an HPLC method to identify and quantify related-substances possibly present into the API. This method has been previously validated by the customer. That is the subject of this article. There are six impurities known (A, B, C, D, E and F) and unknown impurities in this health product when (HPLC) High-Performance Liquid Chromatography is used. Experimental conditions In order to quantify ingredient impurities, an HPLC with MWD (Multi Wavelength Detector) detector was used. Operatory conditions are described below: Instrument: 1100 Agilent Column: Bio-Rad Aminex HPX-87H Flow rate: 0,5 ml/min Flow mode: Isocratic Mobile Phase: water acidified with 7mM of sulfuric acid Injection volume: 20 µl Detection: MWD at 208 nm This method was transferred according to a laboratory protocol prepared by the expert laboratory (laboratory which had performed the validation) and the receiving laboratory (Saint-Vulbas site). To perform this transfer, which is based on validation parameters to be tested, it is necessary to perform different tests such as specificity, sensitivity, accuracy, and repeatability. For all analyses, the API was diluted in water (mg/ml). Each impurity is analysed individually to determine its retention time and to confirm the specificity of the method. The sensitivity of the HPLC is also tested with injection of the API at LOQ level (Limit Of Quantification). Several injections had to be made. All Impurities were spiked at different concentrations in a solution containing the API. In first at LOQ level, in second at specification level (or named 100%, which is the maximum concentration of impurities allowed in the API), and finally at 150%. Three preparations were prepared for LOQ and 150% level, six preparations were prepared for specification level. To transfer the analytical method, the recovery between the spiked quantity and the determined quantity by the analysis concentration of each impurity must be between 70%-130% for LOQ level and 80%-120% for the two other levels. These tests were performed during a week. The stability of impurities at specification level was lastly studied. One of the six preparations is injected every three hours during twenty-four hours. The relative difference of the concentration impurities between the point 0 and the point tested had to be less then 20%. Results First of all, the specificity was evaluated. Each of the impurities is prepared individually and injected into the HPLC system. None of them have the same retention time, as shown on the chromatogram on figure 1, there are no interferences between the different peaks and the resolution is compliant. So the method is specific. Secondly, a standard composed of API was injected at LOQ level. The S/N (Signal to Noise) was superior to 10. Sensitivity was tested according to the FDA pharmacopeia. Thirdly, the accuracy was studied. All recoveries for each preparation were compliant for the specifications. These results are exposed on Figure 2. The method is validated for this parameter. Fourthly, all the RSD (Residual Standard Deviation) were less than 10% for the six impurities, which is the limit set by related-substances. Repeatability of the method is validated too. Finally, the stability of the solutions is fixed at 6 hours because impurity E was unstable in water preparation. Conclusion To conclude, the analytical method was successfully transferred. Indeed, all the results were compliant the acceptance criteria. The analytic method is specific, sensitive, accurate and repeatable for the validated range. Using water for the preparation is easy but the stability of the preparation is limited. Analyses have to be quick in order to give the right result. The analytical method was successfully transferred between the expert laboratory and Saint-Vulbas site. |
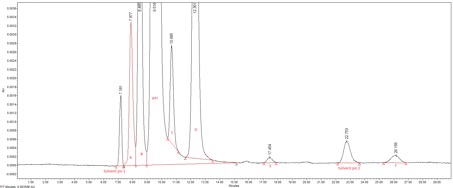 Figure 1: Chromatogram of an API sample spiked with the six impurities known, each impurity can by separated with the HPLC method. It's a specific method for this API. 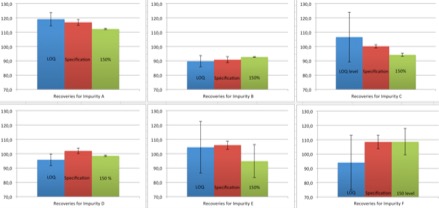 Figure 2: Graphic representation of recoveries of the six impurities. Confidence interval at 95% is added too. Specifications for LOQ are validated because averages are between 70% and 130% of the real values. Averages for specification level and 150% level respect the specifications too. There are all between 80% and 120% of the true values |
|||
|
Siegfried St. Vulbas SAS |
|
|||
ANALYSE & CONTROLE, le MASTER Batiment Bertholet - 22 avenue Gaston Berger 69622 Villeurbanne Phone : 04-72-44-79-88 mail : master-analyse-controle@univ-lyon1.fr |
DIRECTOR Jerome RANDON SECRETARY |
TECHNICAL STAFF Herve DELEPINE Julie BERTRAND Didier FOURNIER |
EDITORIAL OFFICE Editorial Director : Jerome RANDON webmastering : Chahira YAHIAOUI |