
|
2017 Toxicology |
||||
|
Method validation of ethyl glucuronide and ethyl sulfate dosage in urine sample by LC-MS² according to ISO 15189 Srouji Clara |
||||
|
Introduction Ethanol is metabolized less than 0.1% in glucuronide or sulphate conjugates: ethyl glucuronide (EtG) and ethyl sulfate (EtS). These two ethanol metabolites are non-volatile soluble compounds eliminated in urine. Contrary to ethanol which remains detectable for 12 hours in saliva, breath, blood or urine, EtG and EtS can be detected in urine for 3-4 days. Furthermore, EtS, unlike EtG, remains stable in samples contaminated with bacteria. Thus, the determination of these two compounds in biological samples has a particular interest in predicting abstinence from ethanol in alcoholic patients. These two biomarkers can be useful in the procedure of recovering the driving license, in the therapeutic care of alcoholic patients but also to discriminate false-positive results in a post-mortem examination due to ethanol production by fermentation. The aim of this study was to validate a method of determination of EtG and EtS concentrations in urine samples by liquid chromatography coupled to tandem mass spectrometry (LC-MS²). Experimental conditions Material Results The retention times were 2.55 min and 5.25 min for EtG and EtS, respectively. The calibration curves were linear between 73.5 to 11,900.0 µg/L for EtG and 39.2 to 3,060.0 µg/L for EtS. Limits of detection (LOD) and quantification (LOQ) were 24.4 µg/L and 64.4 µg/L for EtG and 15.5 µg/L and 43.6 µg/L for EtS, respectively. Conclusion If precision, cross-sample contamination, LOD and LOQ were successfully validated according to NF EN ISO 15189, accuracy, uncertainty and comparison with other methods have to be performed. If the commercial Chromsystems method is a fast, simple and reproducible method, it presents the drawback to be a closed method where the composition of reagents and the nature of the analytical column are unknown, impeding its potential optimization. This method will be used routinely in the Department of Clinical Pharmacology and Toxicology for the monitoring of chronic alcoholic patients but also in the context of forensic analysis. |
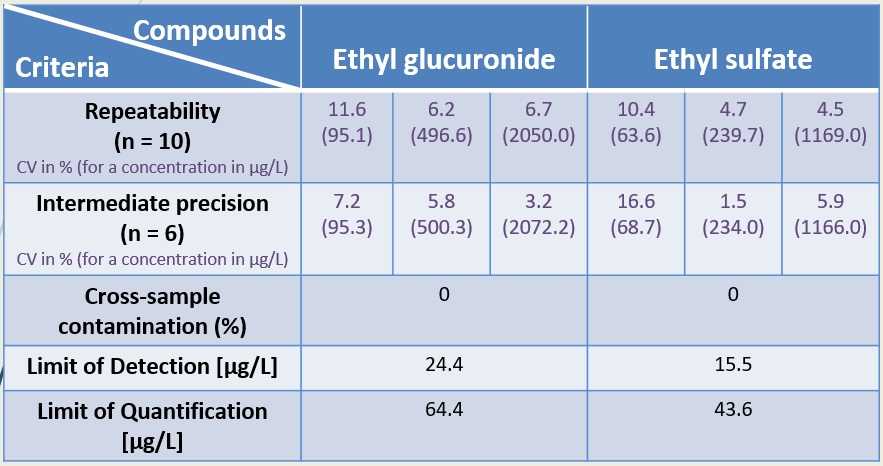 Table 1: Tested Criteria |
|||
|
Service de Pharmacologie Clinique et de Toxicologie - Hôpital Central CHRU de Nancy |
|
|||
ANALYSE & CONTROLE, le MASTER Batiment Bertholet - 22 avenue Gaston Berger 69622 Villeurbanne Phone : 04-72-44-79-88 mail : master-analyse-controle@univ-lyon1.fr |
DIRECTOR Jerome RANDON SECRETARY |
TECHNICAL STAFF Herve DELEPINE Julie BERTRAND Didier FOURNIER |
EDITORIAL OFFICE Editorial Director : Jerome RANDON webmastering : Chahira YAHIAOUI |