
|
2016 Biology |
||||
|
Development of a multiplex method for the proteome characterization of Staphylococcus aureus by mass spectrometry Vanhalle Floriane |
||||
|
Introduction Mass spectrometry is an analytical technique, which generates ions separated by their mass-to-charge ratio (m/z). Currently, this technique is notably applied to characterize and quantify macromolecules such as proteins for system biology or in clinical research. In the ANABIO-MS laboratory (Analyses of Biomolecules by Mass Spectrometry) of the Institute of Analytical Sciences, a quantification method based on liquid chromatography coupled to tandem mass spectrometry (MRM mode) was developed, targeting 700 proteins to study the proteome of a clinical Staphylococcus aureus strain. Experimental conditions Cell pellets of two inactivated Staphylococcus aureus strains were used for the experiment. After extraction, proteins were digested by an endopeptidase (trypsin) to generate tryptic peptides. Trypsin cleaves peptide bonds of the basic amino acid residues lysin and arginin. To simplify the peptide mixture, a MCX solid phase extraction (MCX refers to a cation-exchanged phase) was performed and four fractions of eluted peptides were collected. Each fraction was concentrated under nitrogen flow and finally, peptides were recovered with a water/methanol solvent mixture. Results MRM data were processed with a Skyline platform with which relative information such as peptide retention times, peak areas and fragment intensities can be visualized as graphs and chromatograms (Figure 1). From this analysis, 65.5% of proteins were detected; for proteins characterized in the multiplex method by two peptides, 80.6% were found (but only 57.1% with one) (Figure 2). Thus, it is better to pick up two peptides for each protein instead of one. Conclusion With the developed multiplex method on Staphylococcus aureus on Strain 1, the analysis on the second strain reveals that 65.5% of all proteins have been detected. Among the rest (34.5%), some proteins might not be expressed in this strain or were not detected due to weaker signal-to-noise ratio. Moreover, 80.6% of proteins monitored with two peptides were detected (for only 57.1% for proteins with one). In the near future, new proteins, either from in-house discovery proteomic data or from publicly available proteomic repositories, will still enrich the MRM assay. |
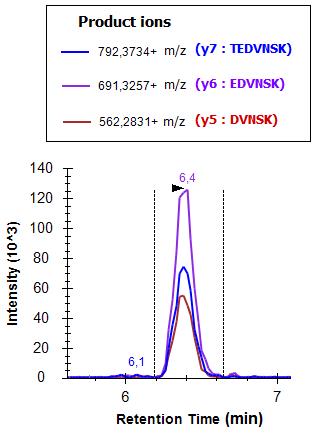 Chromatogram showing the three transitions of a peptide (LTEDVNSK corresponding to a m/z precursor value of 453.2324++) of Staphylococcus aureus, strain 2. The + number refers to the positive charge of the precursor or peptide ion. 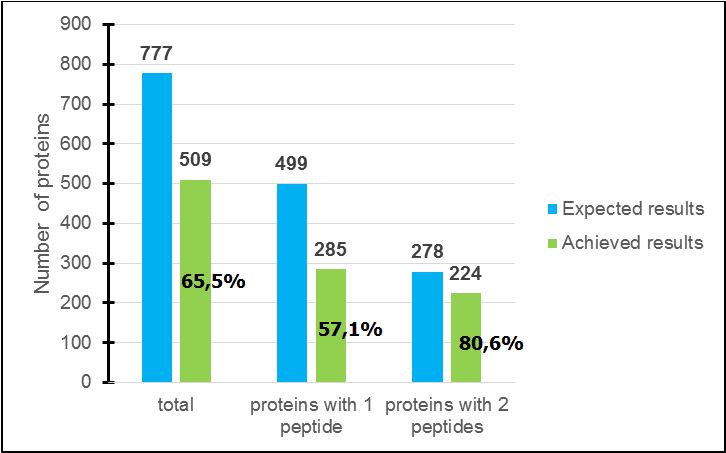 Comparison between expected and achieved results relative to proteins characterized with one or two peptides in the multiplex method. |
|||
|
Institute of Analytical Sciences - ANABIO-MS |
|
|||
ANALYSE & CONTROLE, le MASTER Batiment Bertholet - 22 avenue Gaston Berger 69622 Villeurbanne Phone : 04-72-44-79-88 mail : master-analyse-controle@univ-lyon1.fr |
DIRECTOR Jerome RANDON SECRETARY |
TECHNICAL STAFF Herve DELEPINE Julie BERTRAND Didier FOURNIER |
EDITORIAL OFFICE Editorial Director : Jerome RANDON webmastering : Chahira YAHIAOUI |